Table of Contents
Executive summary
- Cardiovascular disease is the #1 cause of death in modern nations
- Lifetime exposure to circulating LDL is the root cause of cardiovascular disease
- Circulating LDL can be dramatically lowered with safe pharmacological treatments
- For young adults in good health, preventative usage of LDL-lowering therapies eliminates future risk of cardiovascular disease
Abstract
Cardiovascular disease (CVD) is the leading cause of death in the developed world. The progression of cardiovascular disease is fully dependent upon the retention of low-density lipoproteins (LDLs) into the arterial wall, triggering the growth of atherosclerotic plaques. Only certain LDLs, namely those attached to apolipoprotein B (apoB) and apolipoprotein(a) (Lp(a)), tend to become pathologically embedded in the arterial wall in the course of their circulation in the bloodstream. Circulating levels of lipoproteins are largely genetically determined, with minimal influence from lifestyle factors, and modern pharmaceuticals are capable of dramatically reducing LDL levels to those incapable of causing cardiovascular disease in the span of a human lifespan. These pharmacological treatments are safe and well-tolerated, and there is no lower limit at which LDL reduction stops being beneficial. For young adults in relatively good health, aggressive LDL reduction will permanently protect them from developing cardiovascular disease with nearly zero downside. Consequently, cardiovascular disease is effectively a fully solved problem.
Introduction
The claim that cardiovascular disease is solved is at first glance outlandish. In 2020, the CDC ranked heart disease as the most common cause of death in the United States, surpassing even the death toll from all cancers combined.1 How is it possible to claim that CVD is “solved?”
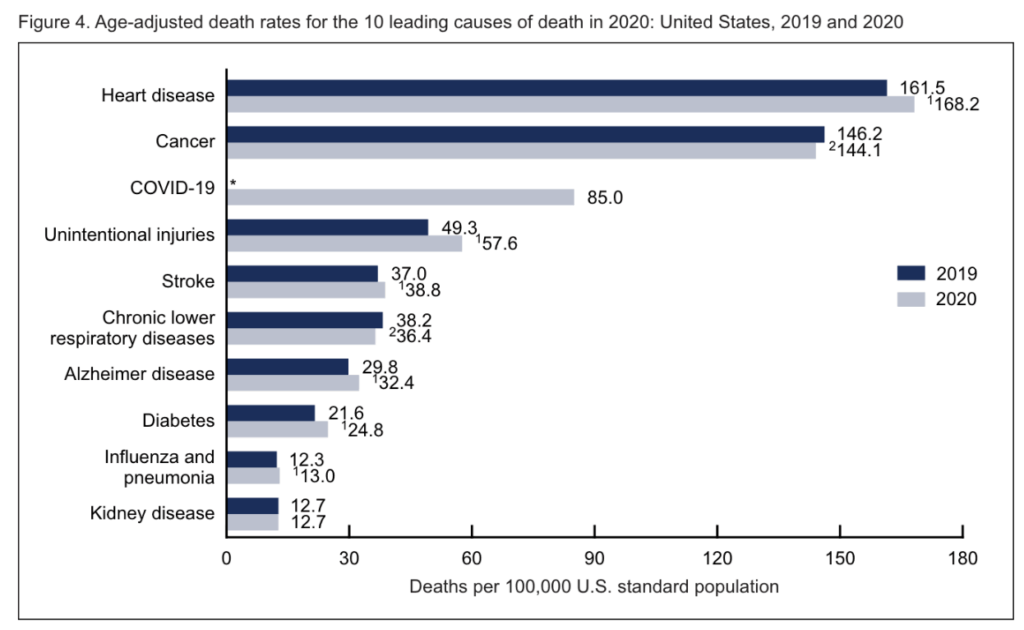
In this article, I will advance the conclusion that cardiovascular disease is, indeed, solved. Half a century of scientific advances, beginning with the development of HMG-CoA reductase inhibitors (also known as statins) in the 1970s and culminating with modern biologics such as PCSK9-neutralizing antibodies have given us the ability to completely and safely ablate the entire causal pathway leading to the development of atherosclerotic disease. Therefore, we are in principle capable of fully eliminating cardiovascular disease from all future generations, as well as for anyone currently living today who is in reasonably good health and has not yet developed irreversible atherosclerotic lesions.
Careful biochemical, epidemiological, and molecular studies have established that cardiovascular disease directly results from the deposition of cholesterol molecules in the subendothelium; this deposition is directly caused by the presence of LDL molecules in the bloodstream; finally, we are capable of reducing circulating levels of LDL well below natural physiological levels. Furthermore, a plethora of theoretical and empirical evidence suggests that reducing circulating LDL levels is purely beneficial to human health. Therefore, the elimination of a cardiovascular disease is essentially a “free lunch” which should be taken advantage of at an individual, and ideally population-wide, level.
This chain of reasoning is not invented from thin air; rather, each step is supported with a tremendous volume of scientific data from the last several decades. The state of scientific consensus has continually evolved toward the direction of more, and earlier, lipid-lowering therapy. The only missing element is a synthesis which takes this reasoning toward its logical conclusion—truly preventative medicine which seeks to eliminate the root cause of cardiovascular disease before it is ever given the opportunity to grow.
Human biology and cardiovascular disease are complex topics, and while I have tried to simplify the discussion to the extent possible, some degree of irreducible complexity remains. The intention of this article is to present a logical, step-by-step argument leading to the conclusions outlined above. Occasionally, short digressions to clarify confusing but essential details have been inserted where appropriate. I ask the reader to kindly tolerate any confusion resulting from my personal deficiencies in writing ability.
Circulating LDL causes cardiovascular disease
Basic pathogenesis
We begin by reviewing the current scientific consensus on the pathogenesis of cardiovascular disease, the history of which has been extensively described in comprehensive reviews.2 Lipoproteins are complex macromolecules which consist of
- an interior with cholesterol and triglycerides,
- a phospholipid outer shell, and
- an apoliproprotein attached to the outer shell.
Their primary function is to transport the interior contents, which are fatty and therefore insoluble in water, throughout the bloodstream, and they are generally categorized according to their size and to the identity of the attached apolipoprotein.
Lipoproteins, especially those smaller than 70 nm in diameter, are continually moving in and out of the arterial wall. Crucially, a small proportion of these lipoproteins, specifically apoB and Lp(a), tend to become trapped inside the subendothelium. This triggers a local, maladaptive, inflammatory response which increases the rate of further lipoprotein deposition, ultimately leading to the formation of an atherosclerotic plaque.
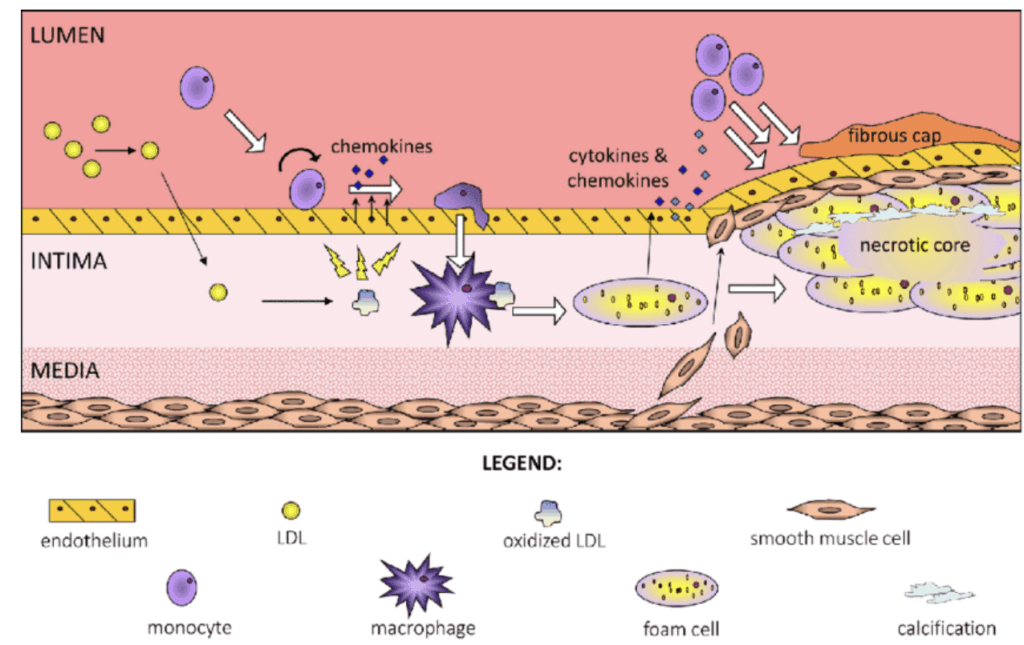
The accumulation of such plaques, known as atherosclerosis, stiffens the arterial wall and restricts the flow of blood, and is the primary cause of cardiovascular disease.
Lipoprotein nomenclature
Before a more detailed discussion of the pathogenesis of cardiovascular disease, it is important to momentarily stop to clarify relevant nomenclature. In order from largest to smallest by diameter, the most common cholesterol-containing lipoproteins are:3
- chylomicrons
- chylomicron remnants
- very low-density lipoproteins (VLDLs)
- LDLs attached to apoB
- LDLs attached to apoB, which is itself attached to Lp(a)
In general, LDLs attached only to apoB are the primary contributor to atherosclerosis; LDLs attached to apoB-Lp(a) are a secondary contributor; finally, all other lipoproteins are relatively minor contributors, aside from perhaps somewhat smaller VLDLs which generally vary in a manner correlated with LDLs attached to apoB.
In this article, the term “LDL” will be used to refer generally to both types of LDLs described above (apoB-only and apoB-Lp(a) moieties). Therefore, taken in conjunction with the fact that these two subcategories of lipoproteins constitute the vast majority of all pathogenic lipoprotein deposits in arterial walls, the term “LDL” can be taken as nearly equivalent to “lipoproteins which cause cardiovascular disease.”
Note that LDL, in this context, is different from LDL-C, which refers to a specific way of estimating serum concentrations of LDL (both subtypes included) from laboratory measurements of high-density lipoprotein (chylomicron and chylomicron remnant) levels and triglyceride levels. Typically, LDL-C is given in units of mg/dL (mass per volume). This should itself be distinguished from “apoB levels” and “Lp(a) levels,” which are measurements of each of the two subtypes of LDL, and may be given in units of either mass per volume or molecule count per volume. The importance of the distinction between measurement by mass and measurement by molecule count will be made clearer at a later stage.
Although seemingly pedantic, the different and overlapping types of lipoproteins along with inconsistent and confusing use of terminology in the scientific literature make it difficult to understand the exact pathways underlying the LDL-based pathogenesis of cardiovascular disease. Clarification of this terminology will therefore clarify the discussion which follows.
Genetic evidence demonstrates LDL causality
Many epidemiological studies have shown strong associations between LDL levels and the risk of developing cardiovascular disease. However, as wide-ranging as these studies may be, they are ultimately observational in nature, and therefore cannot definitively speak to the causal relationship between LDL levels and cardiovascular disease. Similarly, although the exact molecular mechanisms of LDL deposition in the arterial wall and arteriosclerotic plaque formation have been clarified through through studies in vitro and in animal models, these again do not definitively show that circulating LDL is a direct causal factor in the development of cardiovascular disease.
Although noncausal evidence is still quite valuable, we will not discuss epidemiological or mechanistic studies in too much detail—they are by nature highly detailed, often difficult to interpret, and easily challenged. Instead, we will briefly examine the two strongest forms of causal evidence relating LDL to cardiovascular disease.
The first type of truly causal evidence capitalizes upon the inherent randomness of genetic recombination in human reproduction. Statistical techniques such as Mendelian randomization and latent causal variable analysis have demonstrated that genetic variants associated with higher LDL levels are also associated with lifetime risk of cardiovascular disease.4–6 The second type of causal evidence is data from randomized clinical trials testing LDL-lowering therapies, which have consistently shown for multiple decades that drugs which lower circulating LDL levels improve mortality from cardiovascular disease.7 Famously, PCSK9 loss-of-function mutations that accelerate clearance of circulating LDL through the natural LDL receptor (LDL-R) pathway are also associated with up to 90% lower risk of cardiovascular disease, a finding which later motivated the development of the highly successful class of PCSK9 antibody drugs for the treatment of cardiovascular disease.
It is valuable to note that both genetic studies and clinical trials encompass different and independent mechanisms through which LDL levels are varied. For example, the genetic loci identified in genetic studies as having dual relationships with circulating LDL levels and with cardiovascular disease risk are associated with over 50 different candidate genes, and the synthetic pathways of the drugs tested in LDL-lowering clinical trials (HMG-CoA and PCSK9) are broadly non-overlapping. The diversity of the LDL synthesis mechanisms involved in these studies further supports the hypothesis that LDL directly causes cardiovascular disease, rather than merely being an indirect correlate of the true causal mechanism. Otherwise, one would expect that only a specific subset of pathways leading to LDL synthesis would be identified by genomic randomization studies or successful pharmacological drug development.
Taken in conjunction with the tremendous volume of scientific literature elucidating the specific biochemical mechanisms through which circulating LDL becomes embedded into the arterial wall and provokes the formation of atherosclerotic plaques, these lines of causal evidence strongly suggest that higher levels of LDL directly lead to higher risk of cardiovascular disease.
Cumulative LDL exposure determines cardiovascular risk
Going off the premise that circulating LDL levels is the primary causal factor driving atherosclerosis, a natural question to ask is: What is the exact mathematical model that describes the relationship between LDL levels over the course of an individual’s lifetime and their risk of cardiovascular events? Starting with the proposed mechanism where LDL molecules embed themselves over time into arterial walls and trigger subsequent plaque formation, we might put forth two intuitive proposals:
- Cardiovascular risk should increase in proportion with cumulative lifetime exposure to circulating LDL
- Holding cumulative lifetime exposure constant, higher exposure at younger ages is associated with higher risk compared to higher exposure at older ages, because there has been more “opportunity” for atherosclerotic plaques to form around LDL deposits
Although studies of cardiovascular risk in younger-age cohorts are few and far apart, a recent study found support for precisely the two hypotheses outlined above:8
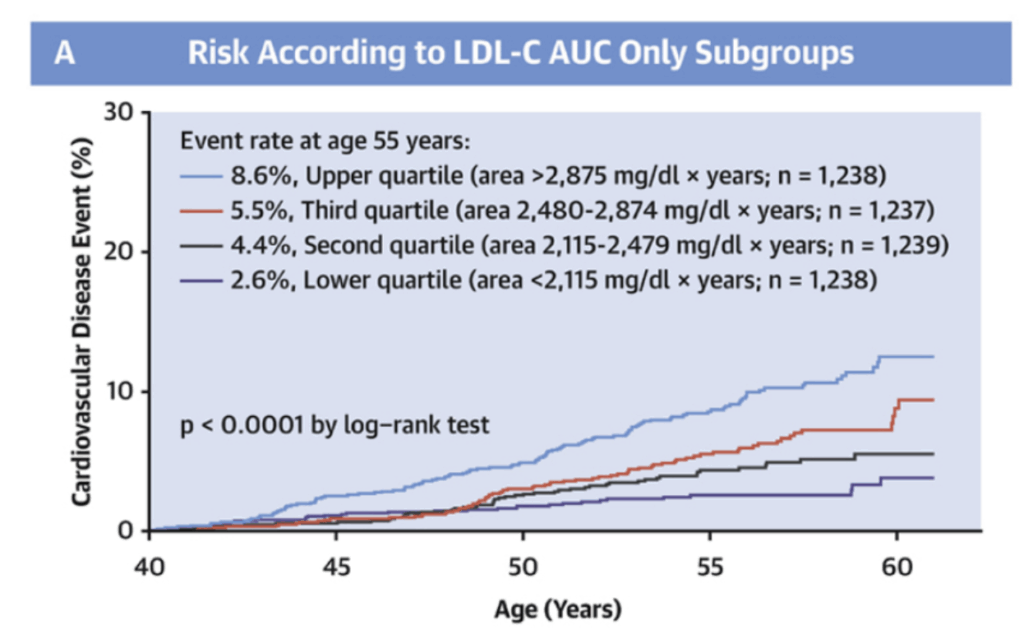
Additionally, subclinical atherosclerosis in middle or old age is commonly observed even at LDL levels considered ‘optimal’ (<70 mg/dL) by standard clinical guidance.9,10 This observation is again consistent with our theoretical model where cardiovascular risk depends on cumulative lifetime exposure to circulating LDL rather than short-term or instantaneous LDL levels.
The concordance of empirical data with our theoretical model has three significant implications. First, it bolsters our confidence that our mechanistic understanding of the pathogenesis of cardiovascular disease is largely correct. Second, it implies that treating elevated LDL in older age is “too little, too late” as the damage from accumulated LDL exposure cannot be easily undone. Third, and perhaps most interestingly, it implies that maintaining extremely low LDL levels well below the ‘optimal’ threshold of 70 mg/dL throughout one’s lifetime has the potential to completely prevent the development of cardiovascular disease.
ApoB particle count is the best metric of LDL levels
So far, we have talked about “LDL levels” in a generic fashion, referring broadly to the levels of deleterious LDL particles in circulation without specifying a particular assay or metric to use. At this juncture, however, it is valuable to take a brief detour to clarify this ambiguity.
Typically, LDL is quantified as a ratio of mass to volume, given in units of mg/dL. However, given our discussion above, it is not necessarily obvious that this is the best way to measure LDL levels in the context of cardiovascular risk. Given our proposed mechanism of CVD pathogenesis, where LDL particles stochastically embed themselves into the arterial wall, one might intuitively hypothesize that:
- larger LDL particles are less likely to flux into the arterial wall, but once they do so, they are more inflammatory, whereas
- smaller LDL particles are more likely to flux into the arterial wall, but once they do so, they are less inflammatory.
Under this model, the pathogenic effect of an LDL particle is not necessarily strongly related to its size. Although the total mass of LDL per volume of blood might still be a reasonably strong predictor of overall cardiovascular risk, another natural candidate might be the number of LDL molecules per volume of blood.
In concordance with the above, recent cohort studies have conclusively demonstrated that the number of LDL particles, as measured by the number of apoB molecules in circulation (recall that each LDL particle is attached to exactly one apoB molecule), is a superior predictor of cardiovascular risk than the mass concentration of LDL in blood.3,11 In practice, LDL mass-per-volume levels and apoB particle counts are often highly correlated, and in patients where the relative proportion of one metric to the other falls within a normal range, both are approximately equivalent for prediction of cardiovascular risk. However, when the relative proportion does not fall within a normal range, then apoB particle counts are a superior predictor of cardiovascular risk (e.g., when apoB particle counts are higher than would be expected based on LDL mass-per-volume levels, then cardiovascular risk is also higher than would be expected from LDL mass-per-volume levels).12 Consequently, whenever possible, reduction of LDL levels (meant generally) should be targeted toward apoB particle counts rather than the typical LDL-C mg/dL measurements reported by standard lipid panels.
Again, the consistency of these empirical findings supports the mechanistic thesis we previously laid out. That being said, for the sake of narrative and conceptual simplicity, we will continue to use the generic, catch-all term “LDL levels” moving forward. However, for any given individual interested in quantifying their own cardiovascular risk, either at baseline or in response to pharmacological or lifestyle intervention, it would be valuable to measure apoB levels in addition to the more standard LDL-C mass-per-volume readout.
Lp(a) and triglyceride-rich lipoprotein levels are secondary risk factors
Before returning to the primary thrust of our argument, we will explore one more brief tangent; although the following details are not crucial to the overall picture, they will aid the reader’s ability to readily understand more fine-grained discussions of cardiovascular risk factors elsewhere.
While LDL levels (specifically, apoB particle counts) constitute by far the primary risk factor for cardiovascular disease and are broadly applicable to almost all people, there are two additional, secondary risk factors worth addressing. These secondary risk factors, namely circulating levels of Lp(a) and triglyceride-rich lipoprotein levels, typically only manifest in specific subsets of the population. In some sense, their root causes are diametrically opposite to each other; Lp(a) levels are almost purely genetically determined and so elevated Lp(a) is essentially due to a poor roll of the genetic dice, whereas elevated triglyceride-rich lipoprotein levels, typically identified through elevated circulating triglyceride levels generally, are characteristic of the metabolic syndrome and diabetic phenotypes which accompany Western diets and the sedentary lifestyles of the modern era.13–15 Both of these risk factors are, in their relevant subpopulations, known to predict cardiovascular risk independent of standard LDL levels, although the mechanistic basis for this independence is not necessarily well understood.
For simplicity, we will devote little further attention to either of these secondary risk factors. In the case of elevated Lp(a), genetic therapies which result in >90% reductions in Lp(a) levels are undergoing clinical trials, and once these therapies are proven and accessible, they will essentially fully eliminate the contribution of Lp(a) to cardiovascular disease, not only in people with genetically elevated Lp(a) but also in the broader population; the problem of cardiovascular risk is then fully reduced to that derived from standard LDL levels, i.e. the case discussed in this article. In the case of elevated triglyceride-rich lipoprotein levels or elevated circulating triglycerides, this is most typically associated with adverse lifestyle or dietary choices. While perhaps unsatisfying from a scientific viewpoint, reasonable lifestyle changes will again reduce the problem of cardiovascular risk to that of standard LDL pathogenesis, and ultimately, it would not be reasonable to expect a ‘one-shot’ solution to both cardiovascular disease and metabolic syndrome. Thus, patients simultaneously at risk for both are advised to first eliminate metabolic disease (either through willpower or through the standard cornucopia of diabetic medications such as metformin, SGLT2 inhibitors, or, more recently, GLP-1 receptor agonists such as semaglutide) before proceeding to the problem of cardiovascular risk.
Pharmacological intervention is necessary for reducing LDL to sub-pathogenic levels
If lowering LDL levels throughout life (especially below the ‘optimal’ threshold of 70 mg/dL given in current clinical practice) is essential to eliminating cardiovascular risk, one might ask: Is there any particular target level of LDL after which further lowering fails to yield cardiovascular benefits, and is this target level achievable with lifestyle changes alone? Interestingly, there seems to be no known ‘lower bound’ after which further lowering of LDL stops yielding cardiovascular benefits, and due to the genetic heritability of LDL levels, lifestyle changes are not sufficient for total elimination of cardiovascular risk. Therefore, for an optimal cardiovascular risk profile, pharmacological solutions must be used to lower LDL levels to sub-physiological levels.
First, studies of twin cohorts or parent–offspring pairs have yielded estimates of the heritability of LDL levels as high as 50%, meaning that up to 50% of individual variation in circulating LDL levels can be theoretically predicted from genetic makeup.16–18 Additionally, the 5th percentile of LDL levels in Americans over the age of 20 is around 75 mg/dL, and in a study of Hadza hunter-gatherers with high amounts of physical exercise and non-Westernized diets, LDL levels largely fell into the range of 55 mg/dL to 80 mg/dL.19,20 Finally, as many as one-third of Olympic athletes exhibit dyslipidemia, as defined by LDL levels exceeding 115 mg/dL.21 Together, these results suggest that for the vast majority of humans, it is impossible to achieve LDL levels much lower than 70 mg/dL even with a theoretically ‘perfect’ lifestyle and diet due to the existence of an individually genetically determined ‘floor’ of at least 50 mg/dL.
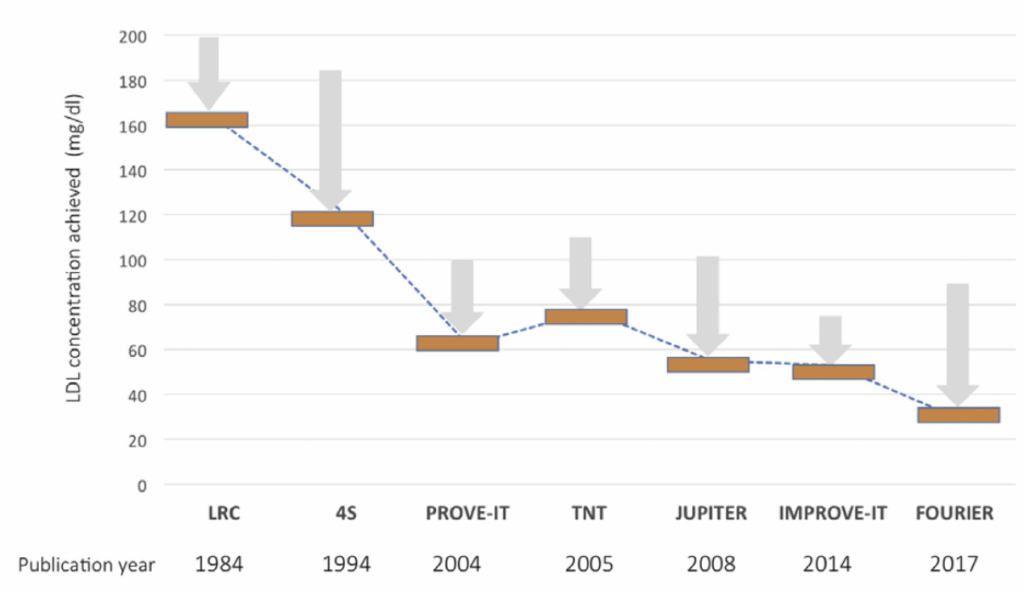
Is there a limit to how far pharmacological lowering of LDL should be taken? As early as in 2005, the PROVE IT-TIMI 22 study showed that cardiovascular risk was reduced by intensive statin therapy lowering LDL below 40 mg/dL.22 Later, in 2014, the IMPROVE-IT clinical trial of ezetimibe in combination to statin therapy showed that cardiovascular risk benefits were seen when lowering LDL levels below 50 mg/dL, and in 2017, the FOURIER trial of evolocumab, an anti-PCSK9 monoclonal antibody, showed benefits to cardiovascular risk when lowering LDL levels below 30 mg/dL.23,24 Finally, a sub-analysis of the FOURIER trial suggested that benefits to cardiovascular risk did not exhibit any plateau even when taking LDL levels as low as 10 mg/dL.25
An important caveat, however, is that these clinical trials typically enroll patients with elevated cardiovascular risk, which typically means older patient populations with high cumulative lifetime exposure to circulating LDL. While these patients continue to show cardiovascular benefits when lowering LDL below 10 mg/dL, that does not imply that it is necessary for a young and healthy individual to necessarily achieve LDL < 10 mg/dL in order to prevent the elimination of cardiovascular disease. Instead, they merely demonstrate that there is no particular lower bound at which LDL reduction should cease, indirectly offering support to our overall quantitative model of the relationship between circulating LDL levels and cardiovascular risk. Additionally, they also demonstrate that modern pharmacological interventions are capable of reducing LDL levels by factors of ten or greater, which would allow people at their ‘physiological baseline’ to potentially attain their target sub-physiological LDL levels.
Unfortunately, it is not clear exactly how far young, healthy adults should lower their LDL levels in order to completely eliminate lifetime risk of cardiovascular disease. No study has ever been conducted to determine the effects of aggressive LDL lowering over a long period of time in early adulthood. However, reasoning logically, if cardiovascular risk is in fact proportional to cumulative lifetime LDL exposure, then at minimum a two- to three-fold reduction below 70 mg/dL is warranted. Taking a rate of atherosclerotic progression which results in subclinical atherosclerosis in later life (which likely still has negative inflammatory and vascular effects, even if it does not immediately precipitate an acute cardiovascular event) and slowing it down by two- or three-fold (targeting 35 mg/dL or 23.3 mg/dL respectively) would theoretically lower the rate of plaque formation to a level such that cardiovascular disease is effectively eliminated from the natural human lifespan.
Achieving extremely low LDL levels is safe
Potential harms of lowering LDL levels are minimal
Although it appears possible to reduce cardiovascular risk to negligible levels through sufficient lowering of LDL levels, doing so would be futile if it significantly increased other health risks. At the very least, the existence of additional risks incurred through lowering LDL levels would cast doubt upon the practical usefulness of targeting low LDL levels; in the most extreme case, they might completely overshadow the cardiovascular benefits of lowering LDL, resulting in overall worsened health and higher all-cause mortality.
There are two specific forms of potential harms that might manifest as a result of aggressively lowering LDL levels:
- first, negative health effects that are intrinsic to low levels of circulating LDL, which might need to exceed some baseline level to satisfy important physiological functions, and
- second, drug-specific adverse effects that manifest at sufficiently high dosages.
Thankfully, neither of these concerns seems to hold substantial weight. Clinical trials of aggressive LDL lowering and data from patients with genetic disorders which dramatically affect LDL production or metabolism consistently demonstrate that lowering circulating LDL levels to < 10 mg/dL has no intrinsic negative effects; in fact, there is considerable evidence suggesting that circulating LDL is little more than a waste product with no biological function in adulthood. Furthermore, long-term studies of all classes of lipid-lowering therapies, including statins, ezetimibe, and PCSK9 inhibitors, show exceptionally good tolerability even at high dosages.
Finally, when fully assessing the risks and benefits of LDL-lowering drugs, a principled approach ought to consider not only negative side effects of lowering LDL but also the potential for any positive side effects. Preventing the deposition of LDL particles into the arterial wall is known to reduce both vascular and systemic inflammation, with pleiotropic benefits ranging from protection of bone mineral density with age to reduction in risk of dementia and cognitive decline.
Admittedly, there is one major, substantiated side-effect of taking a specific form of lipid-lowering therapy: specifically, the risk of ‘statin-induced myopathy,’ i.e., muscle weakness and pain associated with statin therapy.26 However, statin-induced myopathy is somewhat rare: it is reported at a prevalence of 1.5% to 5% in randomized clinical trials, a figure which is an overestimate of the ‘true’ prevalence due to (1) a relatively high base rate of muscle pain in the unhealthy, elderly populations typically enrolled in trials for lipid-lowering therapy and (2) psychosomatic expectation of myopathy caused by lay association between muscle pain and LDL-lowering drugs.27 In any case, statin-induced myopathy ceases upon discontinuation with no known lasting effects, so there is little harm in at least trying statin therapy as a first-line option.
In sum, both empirical and theoretical reasoning support the thesis that there is no level of circulating LDL which is “too low,” and certainly not when taking into consideration the substantial, but not unlimited, degree of LDL reduction which is practically attainable with modern pharmaceuticals. Consequently, the reduction of cardiovascular risk through lipid-lowering therapy can be thought of as akin to a “free lunch.”
Clinical trials show that achieving low LDL is empirically safe
High-quality randomized clinical trials are a compelling source of evidence for the safety of achieving low LDL levels. Although lipid-lowering therapy has been tested many times over the course of the last half-century, we will focus specifically on recent trials which have aggressively lowered LDL levels to sub-physiological levels. Such trials typically use high doses of specific pharmacological therapies to achieve such low LDL levels; therefore, examining the prevalence of adverse effects in patients with the lowest LDL levels simultaneously tests for both forms of potential harms (intrinsic to low LDL levels and intrinsic to the specific drug).
There are at least three recent clinical trials in which intensive lipid-lowering therapy achieved sub-physiological baseline LDL levels for a meaningful subset of patients with a good safety profile:
- Patients in the IMPROVE-IT trial treated with a combination of simvastatin and ezetimibe who achieved LDL levels below 30 mg/dL did not exhibit an increased rate of any adverse events over a 6-year follow-up period.28
- Patients in the FOURIER trial treated with the anti-PCSK9 antibody evolocumab who achieved LDL levels below 10 mg/dL did not exhibit an increased rate of any adverse event over a median of 2.2 years of follow-up.25
- Patients in a pooled study of multiple ODYSSEY trials treated with the anti-PCSK9 antibody alirocumab who achieved LDL levels below 15 mg/dL did not exhibit an increased rate of any adverse events over a follow-up period as long as 2 years except for a slightly greater rate of cataract formation in patients achieving LDL < 25 mg/dL. However, the subsequent ODYSSEY OUTCOMES trial, in which more patients achieved lower LDL levels for a longer duration, did not substantiate any clear connection between extremely low (< 25 mg/dL) LDL levels and cataract formation.29 Given the very slow rate at which lipids are incorporated into the ocular lens, it is likely that the association found in the initial ODYSSEY program was spurious or due to a selection effect.30
Additional studies or subgroup analyses have also conclusively ruled out a role of low LDL levels in insulin resistance or hemorrhagic stroke, both conditions once initially suspected as risks of aggressive lipid-lowering therapy.7 It should actually be noted that these studies actually underestimate the safety of dramatically lowering LDL levels in young, healthy adults. The patient population in these clinical trials is typically older and more diseased to begin with, meaning that they should exhibit inferior drug tolerability at any given dose; moreover, these patients often need high-intensity lipid-lowering therapy precisely because of their existing cardiovascular dysfunction, whereas preventative application of LDL-lowering therapies need not demand such high doses and low target levels of circulating LDL.
In general, the safety of achieving very low LDL levels through pharmacological agents has been studied extensively, with many different treatments and in different medical contexts; it would be impractical to exhaustively cover each and every such study in this article, but detailed reviews doing so already exist elsewhere. However, the overall message is clear: even in the most aggressive treatment regimes which drive LDL levels below 20% of the physiological baseline, no association with detrimental health outcomes has ever been clearly substantiated, suggesting that very low LDL levels, as well as the varied pharmacological agents used in these studies, are both extremely safe.
Genetic evidence suggests that circulating LDL is biologically redundant
In addition to the strong empirical evidence from randomized clinical trials for the safety of achieving low LDL levels, a theoretical argument can be made that circulating LDL is biologically redundant—that, in other words, lowering circulating LDL as low as zero would not necessarily have any detrimental effects. Although it is not actually practically feasible to achieve LDL levels below 1 mg/dL, this argument nevertheless bolsters our confidence that sub-physiological levels of circulating LDL do not pose any risks to long-term health.
In genetic disorders where patients are completely unable to produce any apoB, such as abetalipoproteinemia or APOB-related familial hypobetalipoproteinemia, we observe not only near-zero levels of circulating LDL but also a wide range of pathologies, such as fat malabsorption, neurological symptoms, and ectopic fat deposition.31,32 These symptoms overlap with the side effects of apoB synthesis inhibitors such as mipomersen and lomitapide, suggesting that lack of apoB is the root cause of these problems.33 Clearly, some nonzero degree of apoB synthesis is required for normal physiological function. However, note that these disorders impact apolipoprotein synthesis systematically, including intracellular apolipoprotein synthesis. We can therefore pose the following question: Does circulating LDL play a vital role in cholesterol transport and intercellular signaling, or are all the essential physiological functions of apoB satisfied by intracellular production?
An initial volley of strong evidence in favor of the biological inessentiality of circulating LDL comes from tabulating all known genetic disorders which result in low-to-zero levels of LDL in the bloodstream:
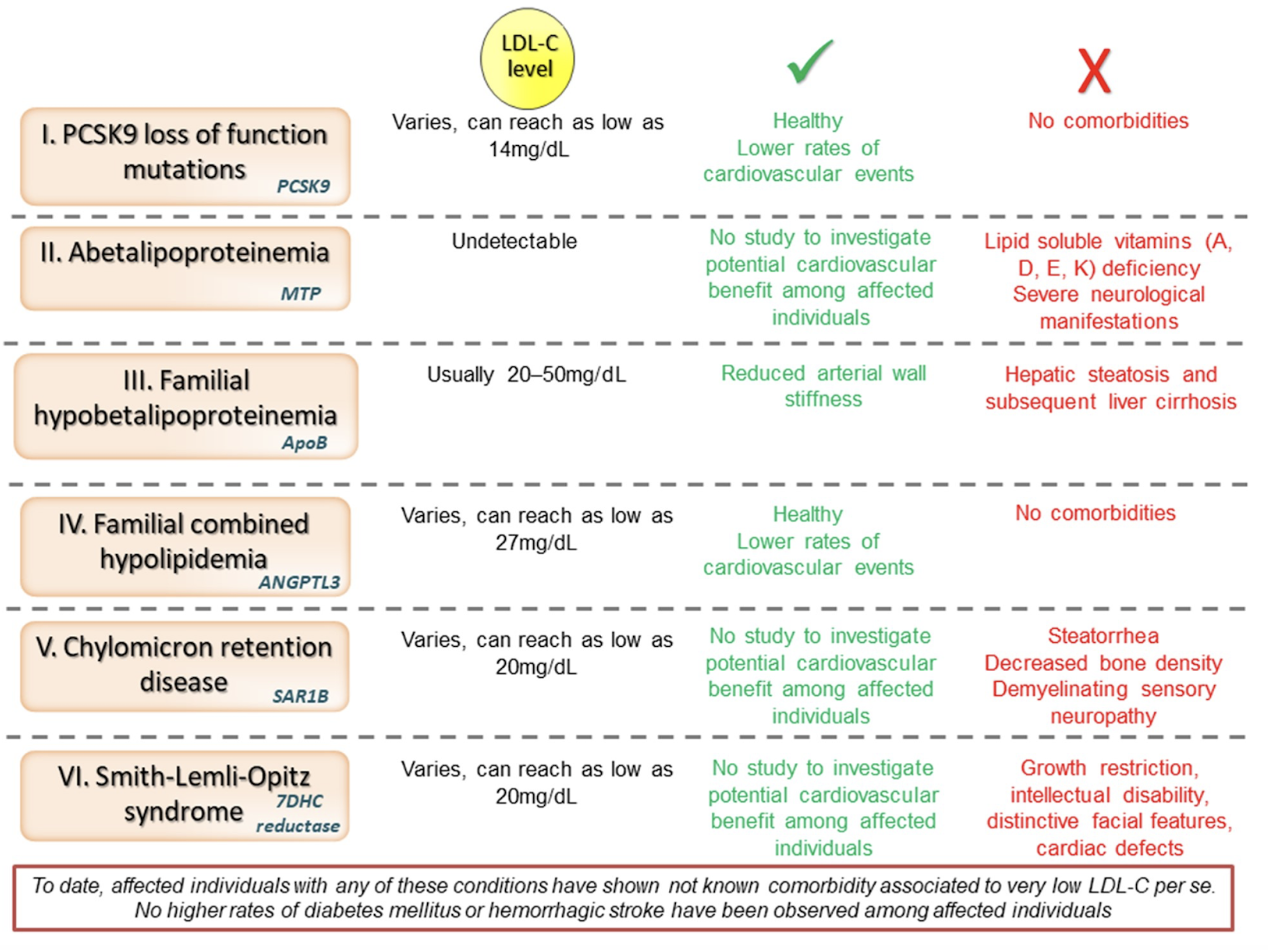
Note that none of these symptoms have any overlap whatsoever with any of the suspected (and eventually disproven) side effects of achieving low LDL levels raised from clinical trial data, such as impaired glycemic control, cataract formation, or hemorrhagic stroke! The complete lack of concordance suggests that the symptoms of impaired apoB production result from an inability to produce intracellular apoB, and that they are completely independent of low levels of LDL in the circulation.
However, an even stronger argument can be made. Recall that approximately 80% of circulating LDL is cleared from the bloodstream after binding to LDL receptors (LDLR) in the liver, and the remaining 20% is cleared via binding to peripheral tissues or alternative pathways:
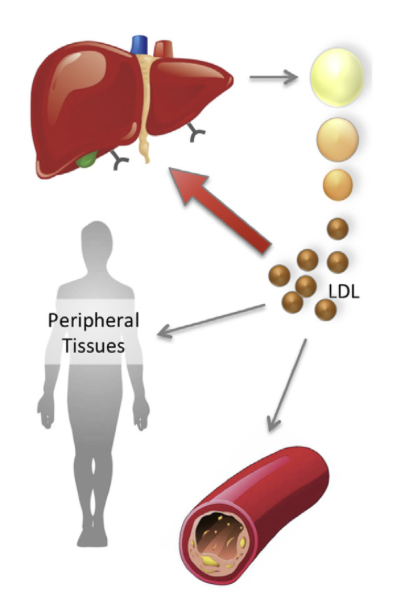
The LDLR-dependent and LDLR-independent clearance mechanisms are mutually compensatory; when there is abnormally high expression of LDLR in the liver, the LDLR removal pathway dominates, with much less LDL being cleared through alternative mechanisms, and when LDLR expression is ablated, all LDL is by definition cleared from the non-LDLR-dependent mechanisms.
In homozygous familial hypercholesterolemia, where LDL receptors are completely absent, circulating levels of LDL are elevated to extremely high levels (>500 mg/dL). As might be expected, patients with this condition develop severe atherosclerosis and cardiovascular disease at young ages. However, interesting, they have normal fetal development, do not exhibit hormonal deficiencies, and have normal neurological function, suggesting that the LDL receptor (through which the bulk of LDL is normally cleared) plays no role whatsoever in these functions, and consistent with the hypothesis that intracellular apoB synthesis is sufficient to satisfy natural physiological demands.7 Much in the same vein, individuals with polymorphisms resulting in higher LDLR activity, which ‘takes away’ circulating LDL from alternative clearance mechanisms, do not exhibit any abnormalities.34 Together, these observations suggest that both the LDLR-dependent and LDLR-independent clearance mechanisms are not essential for intercellular transport or signaling and, consequently, that circulating LDL is akin to a waste product which should be cleared from circulation as fast as possible.
Lowering LDL has many non-cardiovascular benefits
Finally, lowering levels of LDL in the circulation, by virtue of reducing systemic inflammation, has a wide range of pleiotropic benefits not limited to the cardiovascular system. In general, the mechanism through which lowering LDL prevents cardiovascular disease is via lowering the rate of cholesterol deposition into the arterial wall and hence preventing the formation of atherosclerotic plaques; however, beyond the direct effects of arterial plaques upon constriction of bloodflow, which eventually lead to outcomes such as heart failure and stroke, atherosclerosis itself both initiates and is exacerbated by a state of systemic inflammation caused by a pathological immune response targeted at the sites of cholesterol deposition. Accordingly, statin therapy is known to have a robust anti-inflammatory effect.35 Although it is difficult to disentangle the cardiovascular vs. anti-inflammatory benefits of statins from each other, general reduction of systemic inflammation should help slow the advance of age-related pathology, an effect which perhaps explains why statins appear to have a mild protective effect against the onset of cognitive impairment.36
One particularly compelling ‘side’ benefit of taking lipid-lowering therapy appears to be preservation of bone mineral density with age. As early as in 2006, meta-analyses of statin usage suggested that taking statins was associated with higher bone mineral density and lower fracture risk.37,38 Subsequent follow-up studies used both Mendelian randomization and latent causal variable modeling to confirm a causal role of lower LDL on preservation of bone mineral density.5,39,40 Given the extremely high prevalence of osteoporosis in older age, which heightens the risk of severe fractures and increases the rate of hospitalization and surgery, the positive effects of lipid-lowering therapy on bone mineral density should result in a meaningful increase to an individual’s healthspan and overall quality of life.
Taken together with the bulk of evidence suggesting the safety of lipid-lowering therapy, the overall cost-benefit analysis swings markedly toward the side of early prophylactic usage of pharmaceutical agents such as oral statins.
Associations with Parkinson’s disease are either spurious or confounded
For illustrative purposes, we will consider the strength of the claims that statins (the most common form of lipid-lowering therapy) elevates risk for, or exacerbates the progression of, Parkinson’s disease (PD).41 If true, this would be a major strike against the practical applicability of lowering LDL levels as a prophylactic against cardiovascular disease, as cognitive disease results in severely reduced quality of life and is far harder to treat compared to atherosclerotic plaque buildup. How strong is this claim?
One major retrospective analysis of insurance claims in 2017 claimed to find an association between statin usage and the onset of PD.42 However, this finding is likely spurious given that PD is a disease associated almost exclusively with old age, suggesting that it is implausible for changes in a 2.5-year period to meaningfully heighten the risk of incident PD. Another study of Korean health insurance data in 2019 again claimed to find an association between statin usage and PD; however, the claimed association is not present for long-term statin usage, only for short-term usage, suggesting that the result is entirely due to a selection effect.43
Certain studies have also claimed that hydrophilic statins specifically are associated with the worsening of PD.44 Yet this result stands in contradiction to other studies, which instead find lipophilic statins to be associated with PD.34 If we expected one class of statins to show a greater effect on PD progression than the other, it would almost surely be the lipophilic class, which crosses the blood-brain barrier (BBB) far more easily than the hydrophilic class. The total lack of coherence in these results, as well as the lack of concordance with the theoretical prediction, again suggests that these associations are merely spurious.
Finally, vascular inflammation is known to be associated with the onset and progression of PD.45,46 This is consistent with the general understanding of systemic inflammation as a causal factor driving cognitive disease; for example, the majority of patients who die from Alzheimer’s disease are found to have concomitant vascular pathology at autopsy.47 This would predict that statins, which reduce vascular inflammation by preventing atherosclerosis, might actually have a protective role against PD, which is in fact precisely what more recent meta-analyses have found.48,49 There is some minor risk that cholesterol levels in the central nervous system (CNS) are important for neurogenesis or repair of neuronal damage, and that these levels could be depleted by the effect of statins in the CNS; however, even in the worst case, this danger is only limited to BBB-penetrant statins, and the lack of clear associations despite the plethora of patient data studied suggest that any risk induced by BBB-penetrant statins must be quite low.
In sum, although concerns between the risk of PD and statin usage were at some time a point of substantial concern, they were based upon studies with poor methodology which often conflicted each other as well as basic theoretical reasoning, and more up-to-date meta-analyses have in fact identified a protective role for statins in cognitive disease, consistent with our overall understanding of vascular disease and the pathogenesis of cognitive dysfunction. Even if a patient wishes to err on the ‘safe side,’ this concern is entirely mitigated by using non-BBB-penetrant statin therapy (e.g., rosuvastatin).
Given the high prevalence with which statins are prescribed and studied, it is not surprising that spurious associations are often identified, only to be later refuted. In addition, observational studies are especially vulnerable to confounding; for example, statin-treated groups are typically worse off in terms of baseline health than non-statin-treated groups even if baseline factors are controlled for, and when differentiating within statin-treated patients, subgroups which achieve lower LDL levels may essentially be composed of elderly patients with worse overall nutrition. Accordingly, when evaluating claims about the dangers of lipid-lowering therapy, it is important to consider the strength of the supplied evidence in the context of existing knowledge about the beneficial effects of reducing vascular inflammation.
Choosing a LDL-lowering drug regimen
Given that aggressively lowering LDL appears to safely reduce cardiovascular risk, plausibly to such a great extent that the specter of cardiovascular disease might altogether be eliminated, the only missing part is a purely practical matter—the question of precisely which pharmaceutical agents to take to achieve a sub-physiological-baseline level of LDL.
In general, without going through the extensive and detailed history of statin therapy, we will simply note that the state-of-the-art for lipid-lowering therapy is a combination of moderate-intensity statins with ezetimibe.28 Combining low doses of both, rather than using high-intensity statin monotherapy, is believed to achieve comparable LDL reduction at lower risk due to the existence of different metabolic and clearance pathways for each drug. A hypothetical patient might therefore first try low-intensity statins, then moderate-intensity statins, then finally a combination of moderate-intensity statins and ezetimibe, ceasing dose escalation once LDL goals are met.
In regard to the specific choice of statin therapy, it might be slightly preferable to choose a non-blood-brain barrier penetrant statin such as rosuvastatin, in order to hedge against the risk that depletion of cholesterol levels in the central nervous system (typically regulated fully independently from the circulation) might have detrimental effects on cognition. Two additional benefit of rosuvastatin are that (1) it appears to have the lowest rate of statin-induced myopathy and (2) only half-doses are necessary to achieve equivalent LDL lowering for adults with Asian ancestry.26,50
Finally, for those with genetically elevated levels of Lp(a), which appears to be a risk factor functioning independently of circulating LDL, small interfering RNA (siRNA) therapies are currently in development which reduce Lp(a) up to >90% from baseline.51,52 Given the ease of producing siRNAs, these therapies will eventually be broadly accessible at low cost, and are likely advisable for any adult with at- or above-average levels of Lp(a).
Ideally, medications should be taken under the guidance of a licensed physician, although depending on the specific country of residence (or source of importation), lipid-lowering pharmaceuticals such as statins or ezetimibe may be available over-the-counter.
Conclusion
The vast majority of available data suggest that lowering cumulative lifetime exposure to circulating LDL is a safe and practical way to fully eliminate cardiovascular risk. However, if achieving near-total elimination of cardiovascular risk through early and proactive lowering of LDL levels is possible, and if the evidence for this proposition is already present in the scientific and medical literature, why is this not already incorporated into modern medical practice? Two possible reasons are the tolerability of LDL-lowering drugs or the intrinsic safety of achieving extremely low LDL levels; as discussed in the prior section, such concerns are largely overstated, especially in light of the fact that cardiovascular disease supplants all forms of cancer combined as a cause of death in the Western world.
A third and more general reason is that modern clinical practice is by nature responsive rather than preventative; as such, the slow development of atherosclerosis even at ‘optimal’ LDL levels is not intrinsically seen as pathological until arterial calcification reaches a level at which risk for acute cardiovascular events is dramatically elevated. In general, the progressive accumulation of physiological damage over many decades is by default not seen as a disease to be treated in and of itself; for example, the loss of bone mineral density over adulthood, resulting in late-life osteoporosis and frailty, was not seen as a real and treatable ‘disease’ per se until the 1980s.53,54 Fundamentally, the animating question of the medical profession is not “is this treatment expected to have a net positive effect on the patient’s long-term health?” but, rather, “is this patient currently in a diseased state?”
Despite resistance, however, the clinical community is slowly accepting lower and lower thresholds, in terms of both LDL levels and chronological age, for starting statin therapy. The benefits of eliminating cardiovascular disease from greater swathes of the population are too compelling to ignore. However, given the very slow rate of change in medical practice, it will likely be many decades before proactive and dramatic lowering of LDL levels starting in one’s mid-twenties is a widely accepted practice, even though this is the logical and inevitable culmination of over half a century’s worth of scientific research into the cause and progression of cardiovascular disease.
Be that as it may, the sluggard march of clinical practice should not stop individuals from drawing their own conclusions from a careful reading of the scientific literature and taking action to optimize their own health. Health, after all, is a matter of choice, not a mystery of chance.
Postscript
As a direct result of this ‘research’ (really mainly a summarization of existing knowledge), which was performed in February 2024, I began taking 5 mg rosuvastatin daily, then increased my dosage to 10 mg rosuvastatin after several months. My circulating LDL has dropped from approx. 90 mg/dL to 50 mg/dL with zero discernible side effects. Increasing my rosuvastatin dosage to 20 mg daily had no discernible effect, so I am remaining at 10 mg and adding 10 mg ezetimibe every other day. My goal is to reach LDL levels of around 30 mg/dL.
References
↑ 1. Murphy, S. L. Mortality in the United States, 2020. (2021).
↑ 2. Borén, J. & Williams, K. J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr. Opin. Lipidol. 27, 473–483 (2016).
↑ 3. Glavinovic, T. et al. Physiological Bases for the Superiority of Apolipoprotein B Over Low‐Density Lipoprotein Cholesterol and Non–High‐Density Lipoprotein Cholesterol as a Marker of Cardiovascular Risk. J. Am. Heart Assoc. 11, e025858 (2022).
↑ 4. Holmes, M. V. et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 36, 539–550 (2015).
↑ 5. O’Connor, L. J. & Price, A. L. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat. Genet. 50, 1728–1734 (2018).
↑ 6. Pham, K., Mulugeta, A., Lumsden, A. & Hyppӧnen, E. Genetically instrumented LDL-cholesterol lowering and multiple disease outcomes: A Mendelian randomization phenome-wide association study in the UK Biobank. Br. J. Clin. Pharmacol. 89, 2992–3004 (2023).
↑ 7. Masana, L. et al. Clinical and pathophysiological evidence supporting the safety of extremely low LDL levels—The zero-LDL hypothesis. J. Clin. Lipidol. 12, 292-299.e3 (2018).
↑ 8. Domanski, M. J. et al. Time Course of LDL Cholesterol Exposure and Cardiovascular Disease Event Risk. J. Am. Coll. Cardiol. 76, 1507–1516 (2020).
↑ 9. Al Rifai, M. et al. The prevalence and correlates of subclinical atherosclerosis among adults with low-density lipoprotein cholesterol <70 mg/dL: The Multi-Ethnic Study of Atherosclerosis (MESA) and Brazilian Longitudinal Study of Adult Health (ELSA-Brasil). Atherosclerosis 274, 61–66 (2018).
↑ 10. Fern, ández-F. L. et al. Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. J. Am. Coll. Cardiol. 70, 2979–2991 (2017).
↑ 11. Sniderman, A. D. et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 4, 1287 (2019).
↑ 12. Sniderman, A. D. et al. Concordance/discordance between plasma apolipoprotein B levels and the cholesterol indexes of atherosclerotic risk. Am. J. Cardiol. 91, 1173–1177 (2003).
↑ 13. Reyes-Soffer, G. et al. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 42, e48–e60 (2022).
↑ 14. Nordestgaard, B. G. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease. Circ. Res. 118, 547–563 (2016).
↑ 15. Williams, K. J. & Wu, X. Imbalanced insulin action in chronic over nutrition: Clinical harm, molecular mechanisms, and a way forward. Atherosclerosis 247, 225–282 (2016).
↑ 16. Jee, S. H., Yun, J. E., Nam, C. M. & Suh, I. Heritability and Segregation Analysis of the Level of LDL-Cholesterol. Korean Circ. J. 35, 233–239 (2005).
↑ 17. Luo, B. F. et al. Heritability of metabolic syndrome traits among healthy younger adults: a population based study in China. J. Med. Genet. 47, 415–420 (2010).
↑ 18. Lamon-Fava, S. et al. The NHLBI Twin Study: heritability of apolipoprotein A-I, B, and low density lipoprotein subclasses and concordance for lipoprotein(a). Atherosclerosis 91, 97–106 (1991).
↑ 19. Appendix III-A Distributions of Total Cholesterol, LDL Cholesterol, HDL Cholesterol, and Triglycerides in the U.S. Adult Population, NHANES III Data (1988-1994) (Serum). Circulation 106, 3237–3240 (2002).
↑ 20. Raichlen, D. A. et al. Physical activity patterns and biomarkers of cardiovascular disease risk in hunter‐gatherers. Am. J. Hum. Biol. 29, e22919 (2017).
↑ 21. D’Ascenzi, F. et al. Cardiovascular risk profile in Olympic athletes: an unexpected and underestimated risk scenario. Br. J. Sports Med. 53, 37–42 (2019).
↑ 22. Sd, W. et al. Can low-density lipoprotein be too low? The safety and efficacy of achieving very low low-density lipoprotein with intensive statin therapy: a PROVE IT-TIMI 22 substudy. J. Am. Coll. Cardiol. 46, 1411–1416 (2005).
↑ 23. Sabatine, M. S. et al. Rationale and design of the Further cardiovascular OUtcomes Research with PCSK9 Inhibition in subjects with Elevated Risk trial. Am. Heart J. 173, 94–101 (2016).
↑ 24. Cannon, C. P. et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 372, 2387–2397 (2015).
↑ 25. Giugliano, R. P. et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet Lond. Engl. 390, 1962–1971 (2017).
↑ 26. Abed, W., Abujbara, M., Batieha, A. & Ajlouni, K. Statin Induced Myopathy Among Patients Attending the National Center for Diabetes, endocrinology, & genetics. Ann. Med. Surg. 74, 103304 (2022).
↑ 27. Abd, T. T. & Jacobson, T. A. Statin-induced myopathy: a review and update. Expert Opin. Drug Saf. 10, 373–387 (2011).
↑ 28. Giugliano, R. P. et al. Long-term Safety and Efficacy of Achieving Very Low Levels of Low-Density Lipoprotein Cholesterol : A Prespecified Analysis of the IMPROVE-IT Trial. JAMA Cardiol. 2, 547–555 (2017).
↑ 29. Schwartz, G. G. et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 379, 2097–2107 (2018).
↑ 30. Borchman, D. & Yappert, M. C. Lipids and the ocular lens. J. Lipid Res. 51, 2473–2488 (2010).
↑ 31. Burnett, J. R., Hooper, A. J. & Hegele, R. A. APOB-Related Familial Hypobetalipoproteinemia. in GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, Seattle, Seattle (WA), 1993).
↑ 32. Zamel, R., Khan, R., Pollex, R. L. & Hegele, R. A. Abetalipoproteinemia: two case reports and literature review. Orphanet J. Rare Dis. 3, 19 (2008).
↑ 33. Gouni-Berthold, I. & Berthold, H. K. Mipomersen and lomitapide: Two new drugs for the treatment of homozygous familial hypercholesterolemia. Atheroscler. Suppl. 18, 28–34 (2015).
↑ 34. Ference, B. A. Mendelian randomization studies: using naturally randomized genetic data to fill evidence gaps. Curr. Opin. Lipidol. 26, 566–571 (2015).
↑ 35. Satny, M., Hubacek, J. A. & Vrablik, M. Statins and Inflammation. Curr. Atheroscler. Rep. 23, 80 (2021).
↑ 36. Chu, C.-S. et al. Use of statins and the risk of dementia and mild cognitive impairment: A systematic review and meta-analysis. Sci. Rep. 8, 5804 (2018).
↑ 37. Uzzan, B., Cohen, R., Nicolas, P., Cucherat, M. & Perret, G.-Y. Effects of statins on bone mineral density: A meta-analysis of clinical studies. Bone 40, 1581–1587 (2007).
↑ 38. Tang, Q. O. et al. Statins: under investigation for increasing bone mineral density and augmenting fracture healing. Expert Opin. Investig. Drugs 17, 1435–1463 (2008).
↑ 39. Li, G. H.-Y. et al. Positive effects of low LDL-C and statins on bone mineral density: an integrated epidemiological observation analysis and Mendelian randomization study. Int. J. Epidemiol. 49, 1221–1235 (2020).
↑ 40. Zheng, J. et al. The Effect of Plasma Lipids and Lipid-Lowering Interventions on Bone Mineral Density: A Mendelian Randomization Study. J. Bone Miner. Res. 35, 1224–1235 (2020).
↑ 41. Al‐kuraishy, H. M. et al. Pros and cons for statins use and risk of Parkinson’s disease: An updated perspective. Pharmacol. Res. Perspect. 11, e01063 (2023).
↑ 42. Liu, G. et al. Statins may facilitate Parkinson’s disease: Insight gained from a large, national claims database. Mov. Disord. Off. J. Mov. Disord. Soc. 32, 913–917 (2017).
↑ 43. Jeong, S.-M., Jang, W. & Shin, D. W. Association of statin use with Parkinson’s disease: Dose-response relationship. Mov. Disord. Off. J. Mov. Disord. Soc. 34, 1014–1021 (2019).
↑ 44. Lewis, M. M. et al. Parkinson’s disease progression and statins: Hydrophobicity matters. J. Park. Dis. 12, 821–830 (2022).
↑ 45. Rektor, I. et al. Vascular pathology in patients with idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 15, 24–29 (2009).
↑ 46. Yu, C.-C. et al. Vascular Inflammation Is a Risk Factor Associated with Brain Atrophy and Disease Severity in Parkinson’s Disease: A Case-Control Study. Oxid. Med. Cell. Longev. 2020, e2591248 (2020).
↑ 47. Attems, J. & Jellinger, K. A. The overlap between vascular disease and Alzheimer’s disease – lessons from pathology. BMC Med. 12, 206 (2014).
↑ 48. Yan, J. et al. Effect of statins on Parkinson’s disease. Medicine (Baltimore) 98, e14852 (2019).
↑ 49. Bai, S. et al. Statin Use and the Risk of Parkinson’s Disease: An Updated Meta-Analysis. PLOS ONE 11, e0152564 (2016).
↑ 50. Wu, H.-F. et al. Rosuvastatin pharmacokinetics in Asian and White subjects wild-type for both OATP1B1 and BCRP under control and inhibited conditions. J. Pharm. Sci. 106, 2751–2757 (2017).
↑ 51. One dose of experimental therapy reduced lipoprotein(a) more than 94% for nearly a year. American Heart Association https://newsroom.heart.org/news/one-dose-of-experimental-therapy-reduced-lipoprotein-a-more-than-94-for-nearly-a-year.
↑ 52. O’Donoghue, M. L. et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 387, 1855–1864 (2022).
↑ 53. Grob, G. N. From Aging to Pathology: The Case of Osteoporosis. J. Hist. Med. Allied Sci. 66, 1–39 (2011).
↑ 54. Grob, G. N. Aging Bones: A Short History of Osteoporosis. (JHU Press, 2014).
Leave a Reply to Peter McCluskey Cancel reply